


Синдром Кернса-Сейра: фото и симптомы
 Синдром Кернса-Сейра впервые был описан в 1958 г. Это заболевание в большинстве случаев развивается из-за крупных потерь (так называемая «делеция») участков митохондриальной ДНК. Протяженность таких потерь может составлять от 2 до 10 тыс. пар нуклеотидов (сокращенно – п.н.). Однако наиболее распространенной является делеция протяжённостью 4977 п.н. Совсем редко развитие синдрома наблюдается в результате дупликации или точечных мутации.
Синдром Кернса-Сейра впервые был описан в 1958 г. Это заболевание в большинстве случаев развивается из-за крупных потерь (так называемая «делеция») участков митохондриальной ДНК. Протяженность таких потерь может составлять от 2 до 10 тыс. пар нуклеотидов (сокращенно – п.н.). Однако наиболее распространенной является делеция протяжённостью 4977 п.н. Совсем редко развитие синдрома наблюдается в результате дупликации или точечных мутации.
Почти все случаи возникновения описываемого заболевания спорадические. Этот факт объясняется высокой скоростью мутирования митохондриального генома. Существует мнение, что наиболее часто потеря участков ДНК в митохондриях соматических клеток происходят в раннем эмбриональном периоде. В половине случаев при этом имеется также унаследованная от матери дупликация D-петли.
В результате делеции происходит аномальное слияние генетической информации. Гены могут синтезировать РНК, но при этом не способны синтезировать кодируемые ими белки.
Синдром Кернса-Сейра: триада признаков
Как правило, болезнь манифестирует в период с 4 до 20 лет и основывается на триаде признаков.
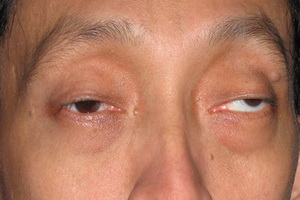
Первой из этой троицы стоит назвать офтальмоплегию, сопровождающуюся птозом верхнего века в купе с ограничением движений зрительных яблок Вторым симптомом данного симптомокомплекса считается прогрессирующая слабость мышц проксимальных отделов конечностей, т.е. тех частей рук и ног, что находятся ближе к месту их начала (например, плечо). И наконец, третий «кит», на котором стоит описываемый синдром – это пигментная дегенерацию сетчатки.
Фото синдрома Кернса-Сейра можно увидеть ниже:
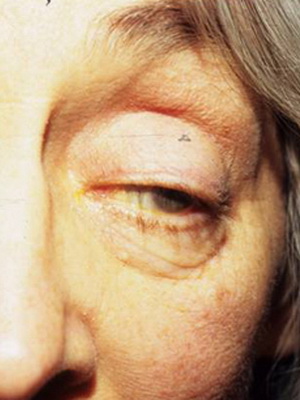
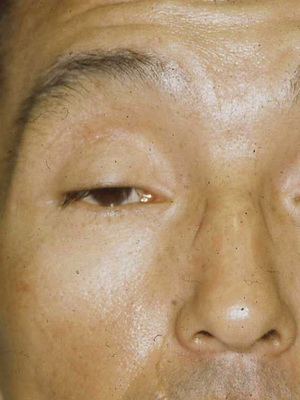
С прогрессированием заболевания появляются и другие проявления: поражается сердце (что выражается в нарушениях ритма и расширении полости желудочков), страдает орган слуха (возникает нейросенсорная тугоухость), затрагивается зрительный аппарат (атрофируется зрительный нерв), а также снижается интеллект.
Смерть больного наступает обычно от сердечно-сосудистой недостаточности по прошествии примерно 10-20 лет от начала недуга. Диагноз, как правило, уточняется при молекулярно-генетическом исследовании биоптатов мышц.
Медицинского лечения описываемого недуга в наши дни пока еще не существует.


 Расшифровка анализа крови
Расшифровка анализа крови Расшифровка анализа мочи
Расшифровка анализа мочи



 Анатомия человека
Анатомия человека Лекарственные препараты
Лекарственные препараты Нарушения обмена веществ
Нарушения обмена веществ Календарь прививок
Календарь прививок Статьи
Статьи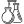 Анализы
Анализы
